衢州市ISO13485医疗器械质量管理体系认证咨询公司,安信达咨询30年专业机构,从策划到落地医疗器械认证专业服务
体系介绍
ISO13485认证有助于企业规范内部管理流程,提升产品质量与安全水平,降低质量风险和合规成本。通过认证,企业的质量管理水平、产品竞争力和客户信任度将得到显著提升。同时,ISO13485认证也是企业申请医疗器械注册证、进行招标投标、拓展海外市场的重要资质支撑。
在衢州市及周边区域,医疗器械产业近年来发展迅速,涌现出众多从事医用电子设备、医用耗材、体外诊断试剂、康复辅助器具等细分领域的企业。衢州作为医疗器械生产经营的重要区域,企业对ISO13485认证的需求日益迫切。
安信达咨询作为成立30年的专业认证咨询机构,为衢州市及周边区域的医疗器械企业提供ISO13485认证全过程咨询服务,帮助企业建立符合标准要求的质量管理体系,培养本地化质量管理团队,确保顺利通过第三方认证审核。
近年来,衢州市及周边地区的医疗器械产业集群效应日益显现,吸引了大量创新型医疗器械企业。这些企业在快速发展的同时,对ISO13485认证的需求也愈发强烈。安信达咨询为这些企业提供从体系策划到认证通过的全流程咨询服务。
体系好处
ISO13485认证能够显著提升企业产品的市场准入能力。取得认证后,企业可凭借认证证书在欧盟、北美、东南亚等主要医疗器械市场开展业务,参与国内大型医疗机构的招标采购,并与国际医疗器械采购集团建立合作关系。
通过体系建立和认证过程,企业能够系统识别产品全生命周期的质量风险点,建立有效的风险预防和应急响应机制。这对于涉及人体健康和生命安全的医疗器械行业至关重要,能够帮助企业降低召回风险和法律纠纷成本。
体系对象
面向已经取得或计划申请医疗器械产品注册证的企业。ISO13485认证证书能够与产品注册形成有效协同,为企业在产品上市、市场拓展、招标采购等环节提供有力的资质支持。安信达咨询在衢州市及周边地区拥有丰富的本地化服务经验,能够为当地医疗器械企业提供及时、高效的认证咨询服务。
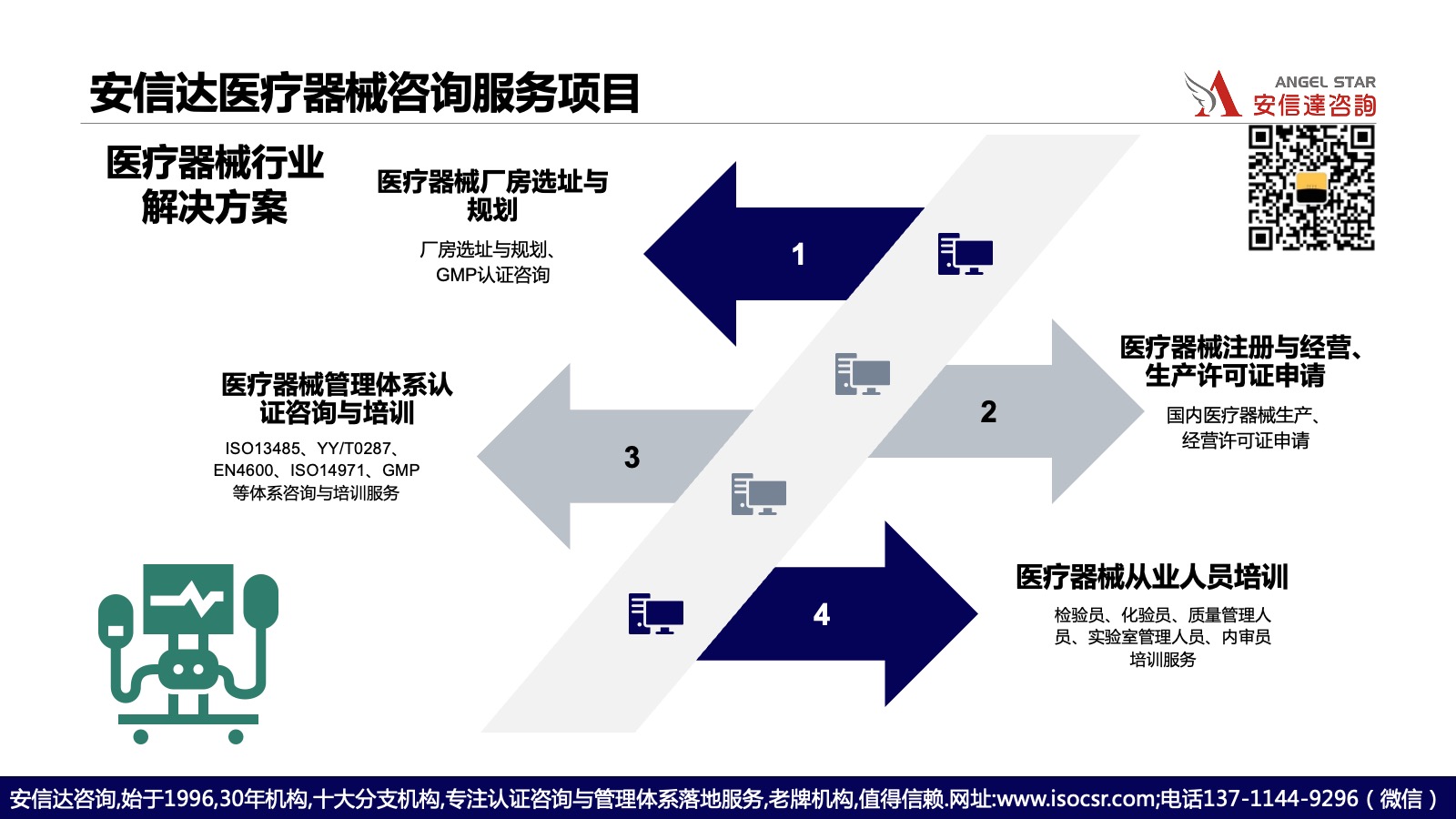
详细的体系建设流程
步骤1:文件编制阶段:在咨询师指导下,企业质量管理团队完成质量手册、程序文件、作业指导书和质量记录的编制工作。文件既要符合标准要求,又要结合企业实际,便于执行和落地。
步骤2:内部审核阶段:试运行达到规定时间后,由企业内审员队伍开展全面内部审核,对体系的符合性、有效性、充分性进行系统性评估,识别需要改进的问题。
步骤3:标准培训阶段:组织企业高层管理人员、关键岗位人员参加ISO13485标准培训,确保相关人员准确理解标准条款的要求和实施要点,建立统一的质量管理语言。
步骤4:认证审核阶段:完成内审和管理评审后,由企业向认证机构提交认证申请。认证机构将派遣审核组对企业进行第一阶段和第二阶段审核。安信达咨询将全程陪同并协助企业应对认证审核。
步骤5:持续改进阶段:取得认证证书后,企业需要持续运行质量管理体系,定期开展内审和管理评审。出现不符合项或客户投诉时,及时采取纠正预防措施,推动体系持续改进。
安信达咨询的服务优势
公司在国内多个主要城市设有分支机构,能够为全国医疗器械企业提供就近服务。同时,安信达咨询建立了完善的项目质量管控机制,包括项目启动前诊断、项目实施过程评审、认证前预审、认证后跟踪等全流程的服务质量保障体系。
安信达咨询始终坚持诚信、严谨、高效、求实的服务宗旨,凭借丰富的专业管理理论、技术和方法,确保客户的医疗器械质量管理体系符合ISO13485标准要求并能有效运作,按计划顺利通过国内外认证机构的认证审核,并顺利取得证书。
安信达咨询在衢州市的本地化服务能力能够为当地医疗器械企业提供快速的咨询响应和现场支持。
常见问题
问:ISO13485认证费用如何
答:ISO13485认证咨询费用需根据企业规模、产品类型、现有管理基础和认证范围等因素综合评估。关于具体的费用信息,建议直接与安信达咨询的业务经理沟通。我们将根据企业的实际情况,提供合理的咨询方案和投资预算。
问:ISO13485认证后需要做哪些持续工作
答:取得ISO13485认证后,企业需要持续开展以下工作:定期进行内部审核(通常每年至少一次全面内审);定期组织管理评审(通常每年至少一次);接受认证机构的年度监督审核;持续维护和更新质量管理体系文件;针对发现的不符合项和客户投诉采取纠正预防措施;持续开展员工培训等。
问:申请ISO13485认证需要具备什么基本条件
答:申请ISO13485认证的企业应具备合法的经营资质,完成相关监管部门的备案或许可手续。企业需要建立并运行符合ISO13485标准要求的质量管理体系,并至少保持三到六个月的有效运行记录,包括内审和管理评审的绩效数据。
问:医疗器械初创企业如何启动ISO13413485认证工作
答:医疗器械初创企业启动ISO13485认证工作建议遵循以下步骤:首先完成企业的合法注册和相关资质许可;然后选择具有医疗器械行业经验的认证咨询机构合作;在咨询师指导下建立质量管理体系框架;完成文件编制和试运行;通过内审和管理评审验证体系有效性;最后申请第三方认证机构的认证审核。
问:ISO13485认证适用于哪些医疗器械企业
答:ISO13485认证适用于医疗器械全产业链的企业,包括医疗器械整机制造商、零部件供应商、原材料生产商、体外诊断试剂企业、医疗器械软件开发商、包装企业等。覆盖的器械范围包括医用电子设备、影像设备、植入器械、一次性使用器械、手术器械、康复辅助器具、医用敷料、医用包装材料等众多细分领域。
问:ISO13485标准最新版本与旧版本有什么主要变化
答:ISO13485:2016是当前最新版本,与旧版本相比主要变化包括:强化与全球医疗器械法规的协调性、增加法规要求的识别与整合条款、强化产品上市后监管和不良事件报告要求、引入基于风险的方法覆盖整个质量管理体系、调整部分术语和文件化信息要求等。
问:ISO13485标准对设计开发有什么要求
答:ISO13485标准对设计开发提出了严格的要求,包括设计策划、设计输入、设计输出、设计评审、设计验证、设计确认、设计转换、设计变更等环节。每个环节都需要形成完整的记录,并确保产品设计满足法规要求、风险可接受、用户需求得到满足。
问:ISO13485标准对风险管理有什么要求
答:ISO13485标准对风险管理提出了明确要求,企业需要按照ISO14971标准对医疗器械全生命周期的风险进行识别、评估、控制和监控。风险管理活动应贯穿于产品设计开发、生产、销售和上市后监管的全过程,并形成完整的风险管理体系文件。
问:申请ISO13485认证需要多长时间
答:实施周期取决于企业现有的管理基础、产品复杂度和资源投入情况。一般而言,从项目启动到通过认证,大致需要四到八个月时间。对于已经通过ISO9001认证或具备一定质量管理基础的企业,周期可以适当缩短。安信达咨询将根据企业的实际情况制定详细的项目计划。
问:ISO13485认证对文件化信息有什么要求
答:ISO13485标准对文件化信息提出了较高要求,企业需要建立完整的质量管理体系文件,包括质量手册、程序文件、作业指导书、质量记录等。文件化信息需要得到有效控制,包括文件的编制、审核、批准、发放、修订、作废等环节,确保现场使用的文件是现行有效版本。
问:医疗器械企业是否必须取得ISO13485认证
答:是否必须取得ISO13485认证取决于企业的产品类别和市场目标。在中国,医疗器械生产经营企业需要符合《医疗器械生产质量管理规范》。在欧盟、美国等海外市场,ISO13485认证是医疗器械注册和市场准入的重要参考依据。对于希望拓展国际市场的企业,取得ISO13485认证具有重要意义。
问:ISO13485认证与医疗器械产品注册有什么关系
答:ISO13485认证与医疗器械产品注册是两个不同的要求,但具有密切关联。在中国,医疗器械产品注册需要符合《医疗器械生产质量管理规范》要求。在欧盟CE认证中,ISO13485是质量管理体系审核的依据之一。在美国FDA审查中,ISO13485认证也有助于证明企业的质量管理能力。
联系电话137-1144-9296 孙老师(微信同号)